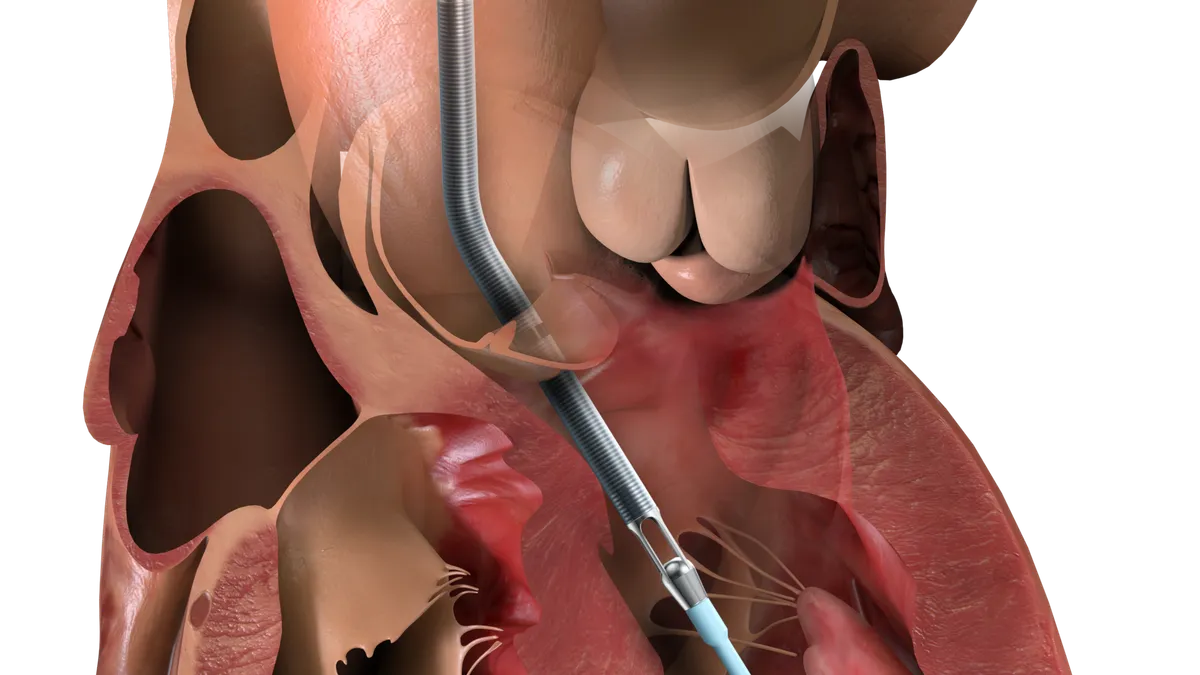
Dive Brief:
- Abiomed completed the post-approval studies requested in relation to the authorization of its Impella heart pumps.
- The Food and Drug Administration now has accepted and closed the post-approval study reports covering the use of the device in high-risk percutaneous coronary intervention, cardiogenic shock, post-cardiotomy cardiogenic shock and right heart failure.
- Abiomed’s studies linked the device to a 22% to 45% improvement in left ventricular ejection fraction, a measure of the volume of blood pumped, after 90 days.
Dive Insight:
Since receiving pre-market approval for Impella in 2015, Abiomed has run five post-approval studies to satisfy the FDA’s request for additional data on the device. The product came under pressure in 2019, when studies linked it to higher costs and more adverse events than balloon therapy. Abiomed weathered the storm, with Impella sales totaling $985 million in 2021, a 22% increase over the prior year.
On Thursday, Abiomed used the FDA’s acceptance of its post-approval study reports as a chance to post an overview of the data it has generated in its Impella pump. Some of the data referenced by Abiomed dates back to 2012, with the most recent publication being an August 2022 retrospective analysis of left ventricular ejection fraction in patients undergoing high-risk percutaneous coronary intervention.
Abiomed posted the overview of the data days after revealing that the FDA has granted 510(k) clearance to its Impella Low Profile Sheath. The sheath has a smaller outer diameter than its predecessor, leading Abiomed to pitch it as a way to ease insertion and removal, reduce procedural steps and help to improve outcomes.
The company also recently shared the findings of a real-world comparison of Impella and an intra-aortic balloon pump run by Yale University. The study linked Impella to improved survival and fewer in-hospital heart attacks and cases of cardiogenic shock.